
AlphaFold3: A foundation model for biology (?)
DeepMind's latest structure prediction model has many new bells and whistles - but what does it mean for the TechBio industry?

Disclaimer: This article is a cross-post of an article I wrote for the Royal Society of Chemistry CICAG Newsletter to appear in July. Therefore, it is assumed the audience already has a basic knowledge of ML, structural biology and AlphaFold2. See Simon’s blog for a technical overview.
Introduction

Since its release in 2021, AlphaFold2 (AF2) has had a transformative impact on structural biology, generating significant excitement in the fields of AI, biology, and drug discovery. Recognising a vast opportunity space, Google and DeepMind spun out Isomorphic Labs to focus on AI-driven drug discovery. Given how competitive the field is, DeepMind are obviously keen to show that they are still on top.
That is presumably the main motivator for unveiling AlphaFold3, which was published in Nature last month along with a webserver to run predictions. The controversial nature (pun intended) surrounding the release of the model has also come to symbolise the transformation of DeepMind/Isomorphic from an open-source altruist to a full-blooded biotech.
On the technical side, the headline news is that AlphaFold3 operates on all-atom coordinates and also uses a diffusion model (a kind of generative model I described in my last article) to build 3D coordinates. The former means it can now generalise to more kinds of biomolecular modelling, including predicting ligands, DNAs, RNAs, modified residues and glycans. The latter means that AlphaFold is now a generative model rather than a predictive one, so it does not always output the same structure and it can sometimes confidently hallucinate bogus outputs (more on that later).
Remarkably, many of the ideas and innovations introduced in AF2, such as equivariance and frames, which have become a defining feature for the research community that the model spawned, were unceremoniously thrown out of the window in favour of brutal simplicity and scale. One could argue that this makes AF3 yet another example of The Bitter Lesson of deep learning (that pursuing more complex and principled architectures is a waste of time if they do not scale). However, this is a model of research that is only possible to pursue at big tech companies such as Alphabet, OpenAI and Meta, for reasons I will explain later.
Unsurprisingly, the model performs very well - strikingly well in the case of protein-ligand interactions, and there is a lot to unpack. This article aims to summarise the main technical distinctions between AF2 and AF3, their capabilities, and discuss the current limitations of the model. Additionally, I will examine the broader implications for the "TechBio" industry. For those seeking an in-depth technical exploration, I highly recommend a peruse of the extensive supplementary methods paper.
The model

The model, while obviously inspired by AF2, has several key differences. It consists of an embedding module, a Transformer-like trunk module that extracts evolutionary relationships and forms a "structural hypothesis," and a final module that generates 3D coordinates. The embedding module is the simplest and transforms discrete residue and atom type information into a continuous latent space.

The first major difference is the second module, called the Pairformer, which generalises AF2's Evoformer to all kinds of biomolecules. In AF2, the Evoformer had a two-track setup: one track took a 2D multiple sequence alignment (MSA) as input, and the other stored representations of pairwise relationships between residues. An attention mechanism at each layer extracted co-evolutionary information from the MSA, passing it to the pair representation track for structural reasoning.
In AF3, the Pairformer retains familiar triangle update and attention mechanisms but applies them to all modalities. Amino acids and nucleotides are tokenized by residue or base, while ligands and glycans are tokenized by atom. Confusingly, this means AF3 has a maximum token length of 5,000 rather than a maximum sequence length. The Pairformer uses a 1D track for sequences and ligand atoms, and a separate MSA module is placed before it to extract evolutionary relationships.

The final major difference is in the module that builds the 3D coordinates from the Pairformer's embeddings. In AF2, this was called the Structure Module. It initialised a "triangular gas" of protein backbone frames at the origin and iteratively predicted rotations and translations on these frames until a structure was formed, with side chain atoms predicted in the last step. Constraints that respected rotations and translations, known as equivariance, were significant here and have inspired much subsequent work.
In AF3, the Diffusion Module replaces the Structure Module. At each step, a random point cloud of atoms is denoised, biased by per-token and atom information. This approach, while much simpler and less “principled”, allows faster iteration and efficient scaling, something DeepMind is uniquely good at. The embeddings from the Pairformer are fed through a few attention mechanisms to predict coordinate updates directly, with absolutely no constraints on equivariance or frames. Although the diffusion process is relatively simple, it is likely challenging to train.
One of the fantastic features of the original AF2 paper was the ablation studies. A good quote that I attribute to Joe Greener of the LMB was that AF2 had “multiple redundant geniuses”, as substantive parts of the model could be removed and still perform nearly just as well. Unfortunately, no such ablation study exists for this paper, making it difficult to extract insights about which of the new ideas contribute to performance.
Being a diffusion model, inference is slow because each of the 200 denoising steps requires an expensive call to the neural network. The example given in the supplementary methods shows inference times of 22 seconds for a 1024-token complex and over 5 minutes for a 5120-token complex, using 16 A100 GPUs. For context, that is more GPUs than I have ever used at once during my PhD, and it only allows for sampling from a model, not training it.
The performance
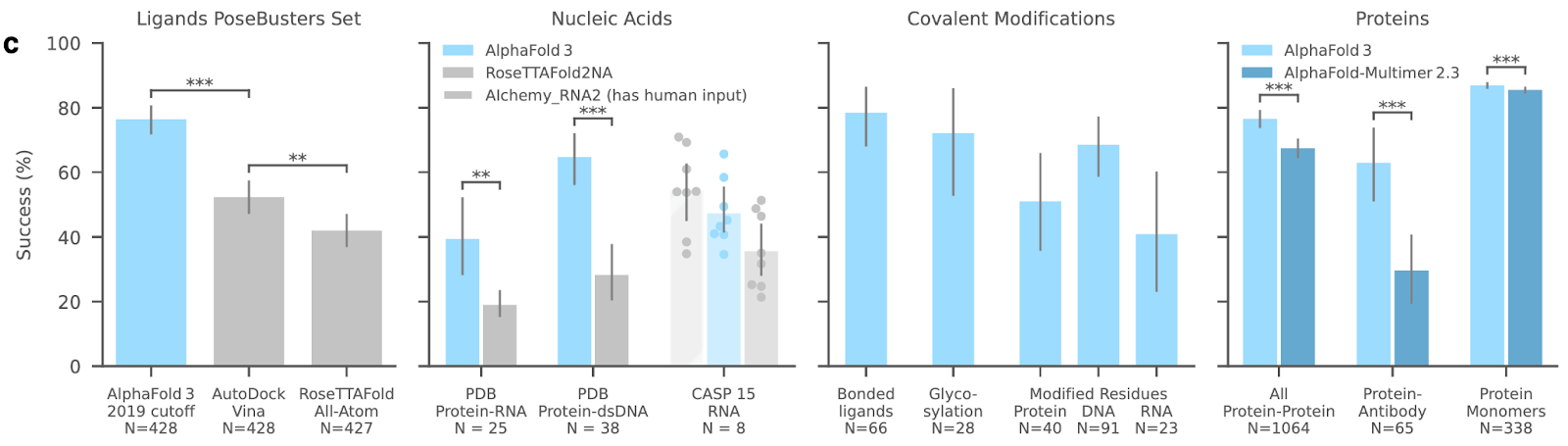
Unsurprisingly, AF3 performs very well in all categories tested, but it is worth clarifying the model’s performance is still an emerging picture, and we should wait for further independent assessments from other groups to be carried out soon. I will keep details on the basic results light here as I want to save writing space for speculation later. Carlos Outeiral (Oxford) has done a great job covering the main results on his blog.
Most interesting for drug discovery is the results on protein-ligand binding, where AF3 comfortably outperforms both AutoDock Vina and RoseTTAFold All-Atom on the PoseBusters set (a test set of recent protein-ligand structures - some of which you can expect to be different from the training data). Even more impressive is that, when binding residues are specified (this is an optional input when running AF3), performance on the PoseBusters docking benchmarking set jumps up to 90.2%, up from 76.4%. (Note: I am only discussing the results on PoseBusters V1 for brevity).

A subject close to my heart is physical realism of generated poses from ML models, thankfully, the paper also reports the success rate of the PoseBusters validity checks (a set of chemical sanity checks developed by Martin Buttenschoen at Oxford). While performance does go down when also considering PoseBusters validity, it does not fall off a cliff like for many other ML methods. This is even more impressive when you consider that they actually remove many physical inductive biases in favour of a simpler architecture, suggesting that fine-tuning your models extremely well is very important here.
This article is already too long, so to grossly summarise the rest of the results I am less qualified to talk about:
They show a marked increase in performance for antibody-antigen complex prediction (although comparison to specialist models is non-existent)
They perform substantially better on protein-DNA complexes than the specialist model RoseTTAFoldNA.
The model can predict covalent ligands, highly relevant to drug discovery.
The model can handle non-canonical amino acids, which is big news for synthetic biology applications and modelling of peptide drugs such as Ozempic.
Give me confidence
An extremely important part of AF2 was not just predicting correct structures, but telling you when you got the right structure (and doing that reliably). This has meant that biologists can either use or safely ignore AF2 models depending on these predicted confidence metrics. Infact, while the infamous spaghetti monster predictions for Intrinsically Disordered Proteins (IDPs) were amusing, the confidence score of AF2 was found to be a near state-of-the-art predictor for such regions, and have further highlighted their relevance in structural biology.

AF3 returns with an even larger smörgåsbord of predicted confidence metrics for every output, measuring both global and local error. While their paper shows that predicted confidence correlates very strongly with accuracy across all modalities, it will take the biological community some time to understand what kind of outputs they can trust.
Limitations
A key question around all deep learning systems is how well they generalise. To oversimplify, deep learning methods perform remarkably well when evaluated on data that is similar to what is seen during training, but sometimes struggle on new data that is very far from the training distribution. Solving the later issue is extremely relevant for drug discovery, where the real value comes from accurately modelling novel targets and/or molecular scaffolds.
Untangling, when and if DL methods trained on biological data generalise has many pitfalls, as similar molecules to your test set may well have been seen in the PDB previously. This was why it was nice to see a figure in the previous AF-latest whitepaper showing that performance went down substantially when modelling novel receptors and/or ligands. Unfortunately, such a plot does not seem to have made its way into the AF3 paper. Additionally, it is hard to determine the generalisability of the model at present since the web server does not allow you to dock your own ligands. Furthermore, a recent study on AF3 found that it was not able to generalise to completely novel metal ion binding motifs not seen during training.

Switching from the non-generative AF2 to the diffusion module in AF3 has also introduced the problem of hallucinations, particularly for disordered regions. This is where the model confidently spits out a totally fictitious output. This can be funny when ChatGPT does this but quite a problem for structural biologists. While the authors say that these regions are usually marked as low confidence, they were obviously more comfortable with the previous set of affairs, and have trained AF3 on predicted structures from AF2 to encourage the model to output the ribbon-like behaviour for such regions.

Key current limitation for all these deep learning-based methods is still their inability to predict dynamics, being limited instead to the static nature of protein structure observed in crystallography. The paper is clear in stating that AF3 has made little progress here, saying that even using different seeds does “not produce an approximation of the solution ensemble”.
Another consideration is the computation cost and the diffusion process makes AF3 is slow. This is particularly a problem in the antibody-antigen binding assessment, where max performance was only reached by taking top-ranking structure from 1000 seeds. It would be an understatement to say that would not be scaleable for virtual screening. The good news this that the server allows you to test antibodies, so we can expect some good benchmarking soon.
Some speculation about what AF3 means for TechBio
Data, data and data
A section with the above title seems to make its way into all my articles, but it is impossible to understate the importance of high quality and diverse data for training these kinds of deep learning models. While the fantastic breakthroughs by DeepMind are often cited, the 50 years of curation and estimated over $12 billion spent on making the PDB are often not. This underscores a massive point: although the ideas introduced by AlphaFold2 have been floating around for years, the progress in tasks other than protein monomer prediction has not been as remarkable. I, and many others, attribute this disparity to the scarcity of available data. (Having said that - I am shockingly impressed that they have done as well as have on protein-ligand complex predicting given that they are limited to the PDB).

Reading the supplementary methods, it is obvious that a substantial amount of effort has been put into the data engineering pipeline. This allows them to quickly collect, merge and pre-process all kinds of biological data, and importantly, rapidly try out lots of different training datasets and splits (they do 4 fine-tuning stages!). Tools like this, along with training code, are rarely published nowadays. The lack of such good open-source tooling for data management here is holding back the research community substantially, so I hope to see a large academic group make one available soon.
A little talked about nugget from the supplementary methods relates to how AF3 was fine-tuned for modelling transcription factors (i.e. protein-DNA complexes). Here, non-structural data was taken from high-throughput SELEX experiments to construct 16k diverse protein-DNA pairs. AF3 was used to make predictions and then fine-tuned on complexes with high predicted confidence. Similarly, the SELEX data was used to construct negative pairs and AF3 was trained to predict idealised monomers of each molecule some distance apart. Although the lack of ablations makes it difficult to to measure the importance of this.
This strategy of using large-scale non structural data, in particular for areas where we are structural data poor such as nucleotides, is quite hot currently. There are a number of startups founded by great ML people moving in this direction, and I predict that we will see Isomporhic try to overcome the data bottleneck with some kind of ultra-high throughput experimental technology such as DELs, deep screening of antibodies or chemotype evolution.
The web server and licence
There is a web server that allows you to run AF3, and I’m sure we can expect to see more independent benchmarking studies coming out in the near future. However, a crucial limitation of the web server is that you're not allowed to dock your own ligands; instead, you can only select molecules from a predefined list (e.g., ATP, NAD). These molecules are all well within the training distribution, so if your protein has a binding site for these molecules, AF3 will likely perform well. Notably, this restriction means that others cannot easily measure AF3's generalisation capabilities to novel scaffolds.
The licence for AF3 is much more restrictive than that for AF2. Commercial use of AF3 is understandably prohibited (unless you pay them a lot for partnerships), and even academics cannot use AF3 predicted structures in any software that predicts ligand or peptide binding (e.g. AutoDock). Additionally, users are prohibited from using AF3 outputs to train their own machine learning models. This is notable because AF2 outputs, available from the AlphaFold Database, have been used for training to easily distill structural knowledge into cheaper models. Moreover, recent improvements in docking generalisation have been achieved by bootstrapping docking models with high-confidence docked poses.
Initially, I wrote a long, detailed analysis of each sentence in the above paragraph, speculating on what each meant for AF3 performance and what DeepMind might be hiding, both positively and negatively. However, on reflection, it is probably just a case that lawyers in the room wanted to have maximum protection over anything IP generating, allowing them to generate maximum revenue from partnerships down the line.
When can we expect an open source AlphaFold3?
Much has been said about the AF3 publication and open-sourcing debate (and, to be honest, I will leave it to those not looking for an industry career!). Regardless, I believe AF3 will eventually be reproduced and open-sourced due to its comprehensive supplementary methods. However, predicting the exact timeline is challenging.
Training AF3 requires significant resources. Based on my estimates, it took around 5,000 A100 GPU days, costing between $100k and $500k for a single training run if reproduced by an academic group (I’m sure Google also calculated the opportunity cost of not training another LLM!). This is many times more than an average PhD student's total GPU usage during their entire graduate career. The actual compute used in model development was likely much higher.
While few academic groups can reproduce AF3, the OpenFold Consortium, led by Mohammed Alquraishi at Columbia, is well-positioned to do this first. They successfully retrained and released the AF2 model with a widely permissible licence. The other confounding factor is that Isomorphic have said they will release an open-source version of AF3 in 6 months (for non-commercial use). As happened with OpenFold, it is likely then that the first PyTorch version of the model will initially use the weights released by DeepMind, with them figuring out how to train from scratch later. At which point, researchers can try and build on top of it.
What does AlphaFold3 tell us about the “AI for drug design” industry?
DeepMind's decision to release AlphaFold3 now is likely driven by multiple strategic goals: securing future partnerships, demonstrating their technological superiority over competitors, and attracting top talent. Isomorphic has already established partnerships with pharmaceutical giants like Eli Lilly and Novartis, securing agreements worth an astounding $3 billion in biobucks—a remarkable figure for such a nascent biotech firm. Given Isomorphic's algorithms are incredibly data-intensive, we can expect further collaborations not only with major pharmaceutical companies but potentially also with startups that possess ultra-high throughput technologies capable of generating the vast datasets they need to keep the music going.
Moreover, Isomorphic's strategy extends beyond external partnerships; the company plans to build its own in-house wet labs to support its data acquisition efforts. This approach is partly driven by the challenges in generalising to novel targets, which has prompted many in the field to focus on amassing data for specific targets. For example, Charm Therapeutics, which has its own co-folding model DragonFold, has invested in developing a wet lab to determine numerous crystal structures, thus creating a protective moat of target-specific data which is only accessible with material venture capital investments.
This signifies a broader trend others have noticed in TechBio from a “first-in-class” to a “best-in-class” approach. Early startups in the space promised AI could discover novel biology, front-loading a large amount of both technological and biological risk by going after novel targets, and have tended to struggle in the clinic. In contrast, newer TechBio companies, riding the hype around AlphaFold and “GenAI”, are collecting large amounts of data for a specific target(s), along with rapid feedback via a lab-in-the-loop (e.g. Prescient Design, LabGenius), hoping to use AI to design better drugs.
Conclusion
As with AlphaFold2, the AlphaFold3 paper is undoubtedly a tour de force of AI and structural biology. This brings me back to the title of this article; is AlphaFold3 a foundation model for biology? The folks at Isomorphic are definitely keen to say so, claiming that they have foundational models for biology and chemistry that work well in tasks beyond the training problem, such as protein design and affinity prediction. Ultimately, we have yet to see any of this publicly and there is still a massive question mark around generalisation, so the ball is still in their court to some degree.
To be fair, DeepMind's remarkable consistency in leading AI-driven structural modelling is particularly striking, given that AF2 was first developed over four years ago. While the recipe for their success is not exactly a closely guarded secret, it does require a unique combination of infrastructure, talent, engineering, and scale - a challenge to replicate without serious investment, which is impossible in academia. Which brings me to another striking point: despite billions of dollars of venture capital investment in "AI for drug discovery," only one industry player, Isomorphic Labs (another Google company), has successfully replicated the kind of team and infrastructure necessary to achieve something like AF3.
Finally, as Derek Lowe pointed out in his blog, structure is not everything, and so far we have yet to see if AF3 improves over other strategies for hit-finding, and if so, at what cost. AF3's capabilities currently focus on binding, but the translation from binding to biological function can be complex. For some targets, binding might be an adequate predictor of function, but for others, it might not suffice. It will take holistic and multi-scale modelling of biology for Isomorphic to truly “solve drug discovery” with AI. Although a quick glance at their jobs page suggests they might be moving in that direction.
I would like to thank Leonard Wossnig, Simon Barnett, Chaitanya Joshi and Helen Cooke for their help in reviewing this article. I would also like to thank Max Jaderberg and Mari Barlow for their help in obtaining permission to reproduce figures and sharing the Press Pack. I would also like to thank my supervisors, Professors Pietro Lio and Sir Tom Blundell, for their guidance and support.
Great write up.